Fold tree:折叠树
Fold tree:折叠树
# Fold tree:折叠树
# 引入
# 内部坐标
如果打开一个 PDB 文件查看,会发现其中包含了所有原子在三维空间中的 (x, y, z) 坐标信息。当想移动一个蛋白质时,可以改变每个原子的三个坐标。换句话说,每个原子具有三个自由度。然而,三个自由度的计算来说太复杂了。
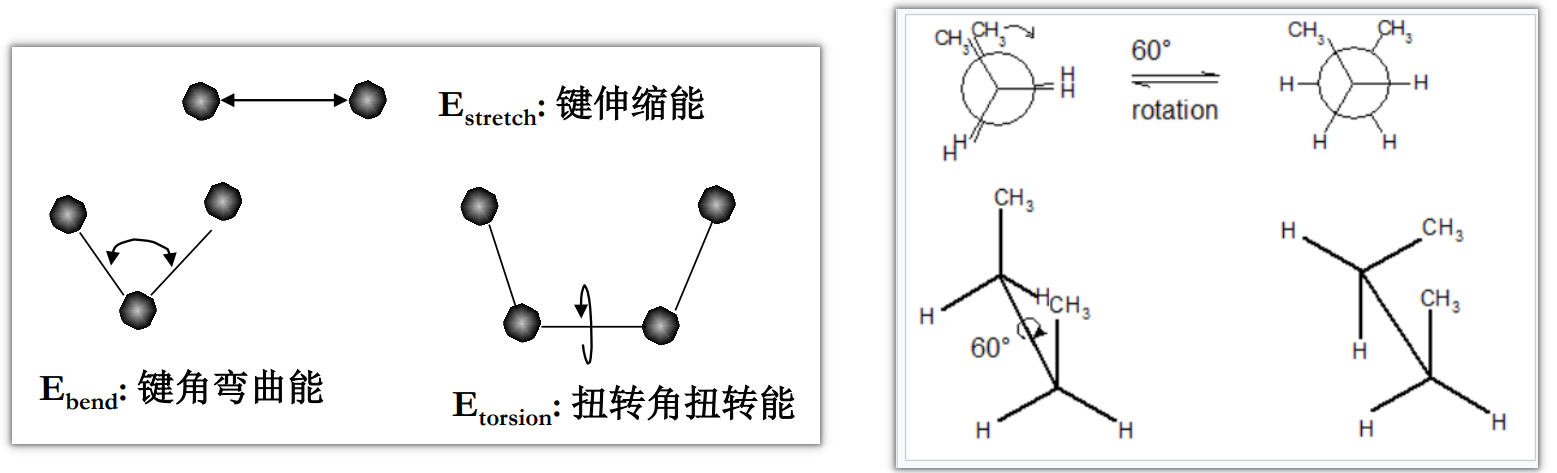
为了简化问题,Rosetta 改用内部坐标。可以想象,定义一个原子的另一种方式是通过它与相邻原子之间的键长、键角和扭转角。 当使用残基的键长、键角和扭转角值,而不是 x, y, z 坐标值来描述一个原子时,就在使用它的内部坐标。
 为什么使用内部坐标:
在内部坐标的世界中,物体是通过改变键长、键角和扭转角来移动的,因此仍然有三个自由度。然而,这些自由度的重要性并不相同。对于大多数蛋白质建模来说,键长和键角几乎不会发生变化 —— 实际上可以认为它们是固定的。 运动主要通过扭转角的变化来实现。因此,将每个原子的自由度从 3 个减少到 1 个。此外,对于某些原子(比如大多数氢原子),它们的扭转自由度被其化学环境固定。对于这些原子,自由度从 3 个降为 0 个。
为什么使用内部坐标:
在内部坐标的世界中,物体是通过改变键长、键角和扭转角来移动的,因此仍然有三个自由度。然而,这些自由度的重要性并不相同。对于大多数蛋白质建模来说,键长和键角几乎不会发生变化 —— 实际上可以认为它们是固定的。 运动主要通过扭转角的变化来实现。因此,将每个原子的自由度从 3 个减少到 1 个。此外,对于某些原子(比如大多数氢原子),它们的扭转自由度被其化学环境固定。对于这些原子,自由度从 3 个降为 0 个。
# 杠杆臂效应(Lever Arm Effect)
当在一组特定残基中改变扭转角,但将其余部分设置为不动时,会发生什么?来看下面这个简单的示意图:
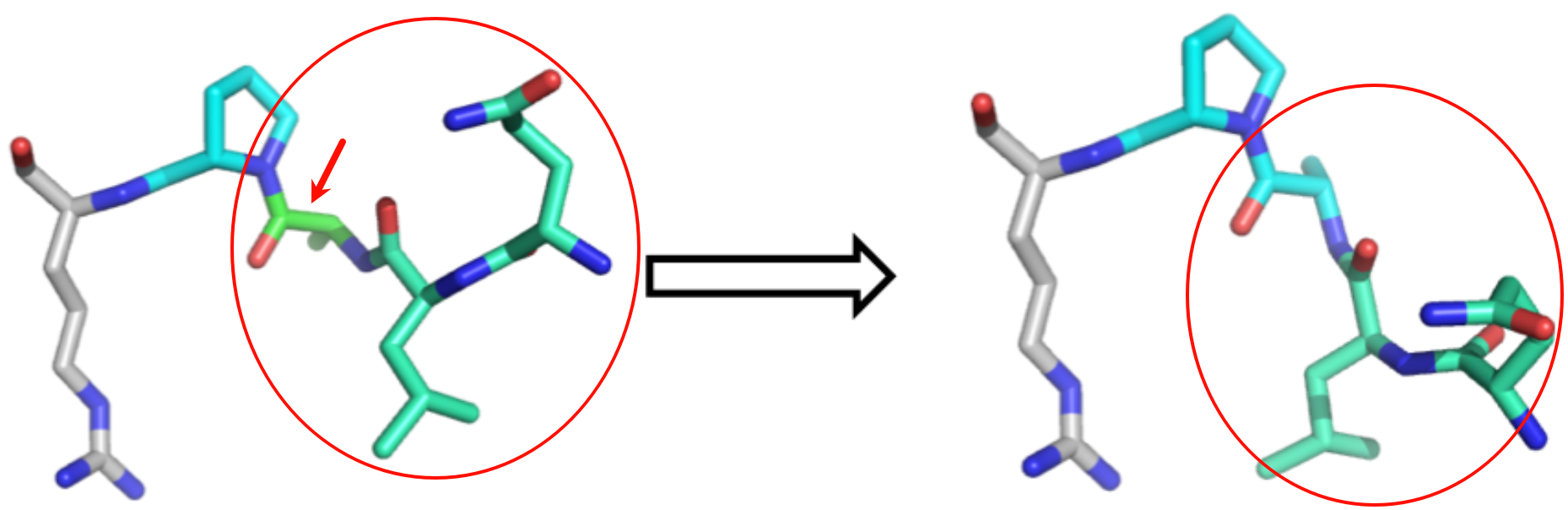 可以看到,为了改变青色残基中的一个特定扭转角,所有在该位置右侧的原子都必须一起移动。换句话说,尽管这些原子“彼此之间”是固定的,但它们仍会一起移动,以适应下游青色残基的变化。
可以看到,在蛋白质某个位置的一个扭转角发生微小变化,就能导致蛋白质其余部分发生巨大的位移。这就是所谓的杠杆臂效应(lever arm effect)。
使用 Fold Tree 一定程度可以避免这件事情。
可以看到,为了改变青色残基中的一个特定扭转角,所有在该位置右侧的原子都必须一起移动。换句话说,尽管这些原子“彼此之间”是固定的,但它们仍会一起移动,以适应下游青色残基的变化。
可以看到,在蛋白质某个位置的一个扭转角发生微小变化,就能导致蛋白质其余部分发生巨大的位移。这就是所谓的杠杆臂效应(lever arm effect)。
使用 Fold Tree 一定程度可以避免这件事情。
# Fold Tree 使用
使用 Fold Tree 来控制蛋白质各部分之间的相对运动。折叠树是一种告诉 Rosetta 给定结构中残基之间连接关系的方式。它定义了哪些残基是上游(upstream) 或父节点,哪些是 下游(downstream) 或子节点。通过一些例子来进行说明。
在这个示例中,将使用冠状病毒核衣壳蛋白(nucleocapsid)N 端部分的结构。
打开 inputs 目录下提供的 capsid.pdb 文件。会看到该结构具有一个较长的、无结构的 N 端,而其余结构主要为 β-折叠片。目前知道 N 端区域是无序的,因此在处理 PDB 文件之前,只想对这部分进行松弛(relax)。
cps_relax1.xml 是一个为提供的 Rosetta 脚本,它使用 MoveMap 对结构中的第 1 到 20 个残基进行松弛处理:
<MoveMap name="part">
<Span begin=1 end=20 bb=1 chi=1/>
</MoveMap>
2
3
它还使用默认的 Rosetta 折叠树,该树显示在 inputs 目录下的 caps_tree1.ft 文件中:
FOLD_TREE EDGE 1 133 -1
这可以读作:“从第 1 个残基到第 133 个残基的一条边,是一条共价连接边(-1)”。
从链 A 的第 1 个残基开始,一直到该链的最后一个残基(本例中为 133)。因此,任何对上游残基(靠近 N 端的残基)的运动,都会导致下游残基(靠近 C 端的残基)发生运动。这个链是一个连续的、不被打断的残基簇,这些残基通过共价键以线性方式连接。称这个簇为一个 EDGE(边)。-1 表示 EDGE 中的残基通过共价键连接。
在 fold_tree 文件夹中,使用以下命令运行脚本,仅对第 1 到 20 个残基进行松弛处理,而不改变其余结构的主链:
$> <path_to_Rosetta_directory>/main/source/bin/rosetta_scripts.default.linuxgccrelease -in:file:s inputs/capsid.pdb -parser:protocol inputs/caps_relax1.xml -out:prefix test1_
现在将原始结构与输出进行比较(例如,在Pymol中对齐它们)。可以看到n端和原来的结构相比有了很大的变化。如果只对齐n端部分,可以看到蛋白质的其余部分已经剧烈地移动,以适应小20个残基n端的变化。
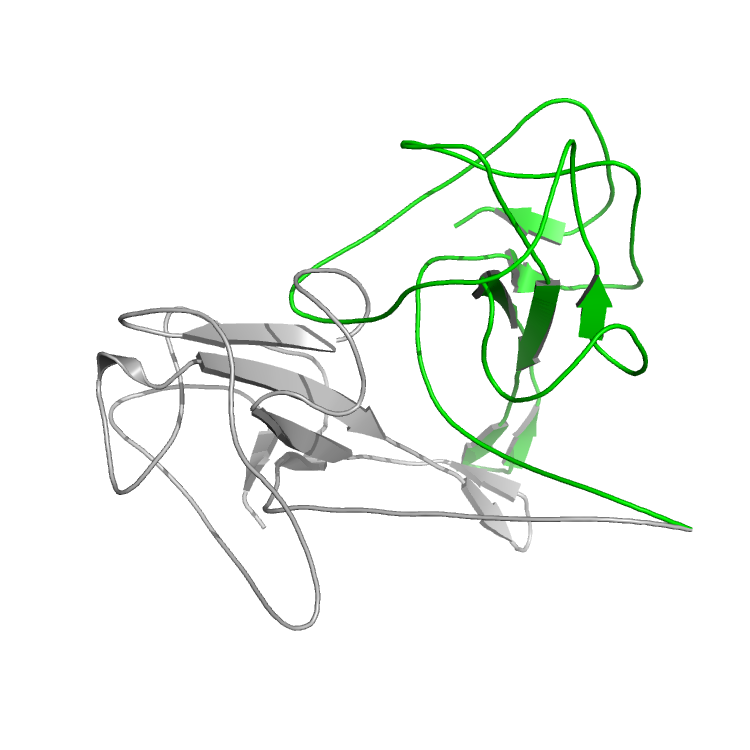 原始结构显示为灰色,test1 的结构显示为绿色。
现在,当交换上下游的位置,也就是告诉 Rosetta,运动应该从 C 端传播到 N 端时会发生什么。这在
原始结构显示为灰色,test1 的结构显示为绿色。
现在,当交换上下游的位置,也就是告诉 Rosetta,运动应该从 C 端传播到 N 端时会发生什么。这在 caps_tree2.ft 文件中显示如下:
FOLD_TREE EDGE 133 1 -1
可以看到,折叠树是有方向性的。
现在对 capsid.pdb 结构运行一次松弛,但这次使用新的折叠树:
$> ../../../main/source/bin/rosetta_scripts.default.linuxgccrelease -in:file:s inputs/capsid.pdb -parser:protocol inputs/caps_relax2.xml -out:prefix test2_
查看输出结构 test2_capsid_0001.pdb,并将其与 capsid.pdb 和 test1_capsid_0001.pdb 进行比较(两个输出文件均位于 outputs 目录中)。会发现,这次蛋白质中的运动小得多。这是因为现在 C 端部分处于变化位置的上游,因此不会因 N 端的运动而发生位移。
 原始结构显示为灰色,test2 的结构显示为青色。
因此,如果蛋白质具有非常灵活的 N 端,在运行过程中会发生较大运动,建议更改折叠树。
原始结构显示为灰色,test2 的结构显示为青色。
因此,如果蛋白质具有非常灵活的 N 端,在运行过程中会发生较大运动,建议更改折叠树。
# 更复杂的折叠树
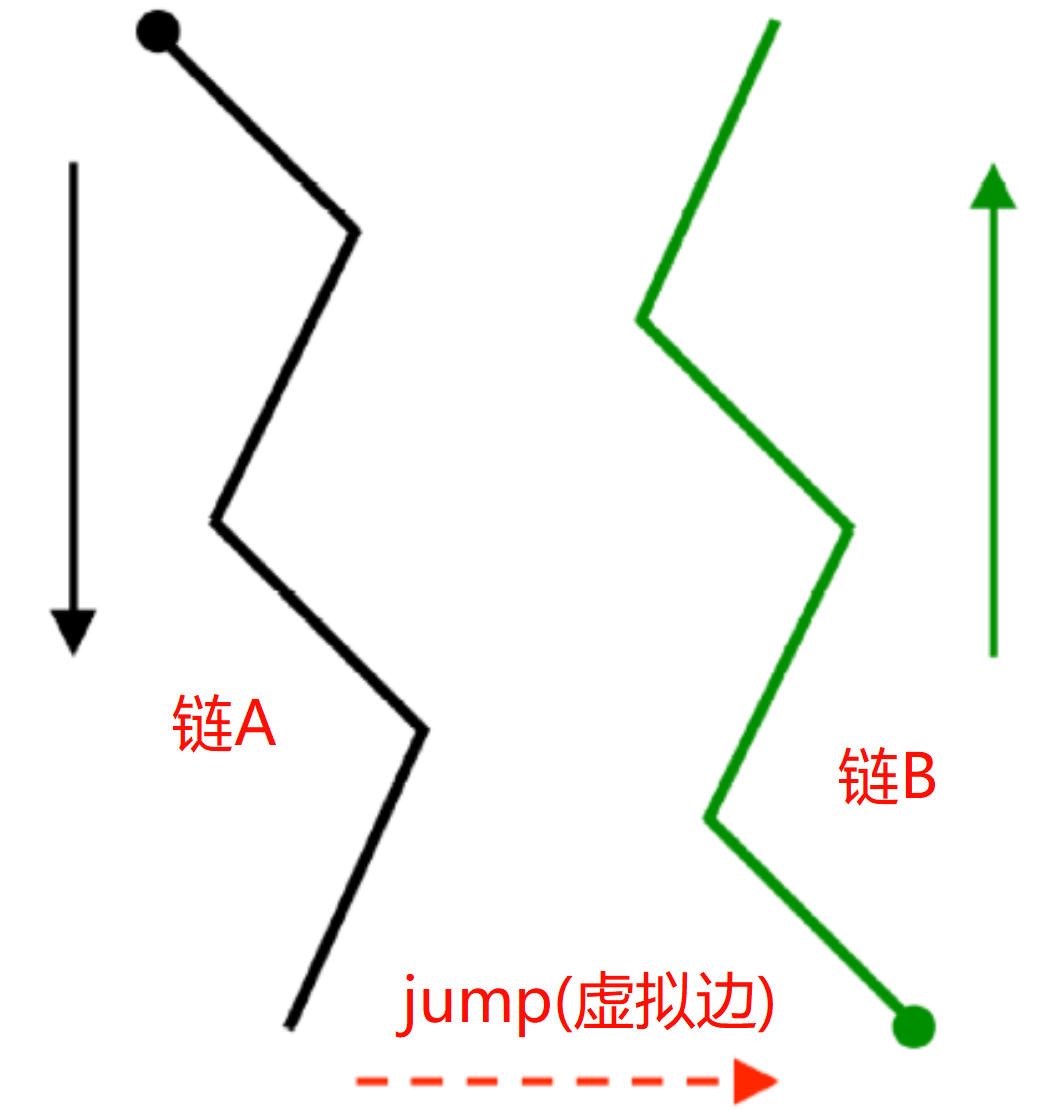
FOLD_TREE EDGE 1 76 -1 EDGE 76 77 1 EDGE 77 152 -1
可以读作:“从第 1 到第 76 个残基是一条蛋白质边(-1);从第 76 到第 77 个残基是第一个跳跃边(1);从第 77 到第 152 个残基是另一条蛋白质边(-1)” 可以看到总共有三条 EDGE:
- 第一条应该很熟悉(和第一个例子一样,就是一条链),它表示从残基 1 到残基 76 是通过共价键连接的(也就是链 A)。
- 第三条做的是同样的事情,但对应链 B。
- 现在来看第二条 EDGE。会发现它从链 A 的最后一个残基(76)连接到链 B 的第一个残基(77)。这不是共价连接! 但由于 Rosetta 使用的是内部坐标(即扭转角),而不仅仅是三维坐标,因此结构中的所有部分必须以某种方式连接起来。如果没有共价连接(就像我们这个例子),链之间就必须通过一个我们称为“跳跃(JUMP)”的虚拟连接来连接。 跳跃也是一条 EDGE。在折叠树中,跳跃用正数表示。第一个跳跃编号为 1,后续跳跃按顺序递增。